
![]()
![]()
![]()
![]()

La vida en una burbuja
Ser Humano

En mis clases de inmunología suelo preguntar a mis alumnos si han visto la película El chico de la burbuja de plástico. Sin embargo, ninguno hasta este momento ha visto el drama protagonizado por John Travolta, de 1976, ni la versión cómica protagonizada por Jake Gyllenhaal en 2001. Estas películas nos hablan de un chico que vive atrapado en una burbuja de plástico, pues carece de sistema inmunológico y por lo tanto exponerse a bacterias, virus y cualquier agente infeccioso podría significar su muerte. Así pues, el personaje principal de ambas cintas vive su infancia y juventud aislado de toda la gente, hasta que llega el amor, esa fuerza que todo lo puede, entonces abandona su aislamiento para ir en busca de la mujer de sus sueños… Esta película no es mera ficción, está basada en historias reales, como la de David Vetter.
David Vetter era un niño que nació con una enfermedad conocida como “inmunodeficiencia combinada grave”, una enfermedad que afecta el desarrollo de las células del sistema inmune [a], que es el que nos ayuda a combatir infecciones y diversos padecimientos. Para darnos una idea de lo seria que es, en el SIDA se reduce el número de unas células del sistema inmune llamadas linfocitos T CD4, lo que lleva a los pacientes a enfermarse con facilidad; en el caso de la inmunodeficiencia combinada grave, el daño al sistema inmune es aún mayor, ya que el paciente carece de muchos otros tipos de células, por lo que suelen fallecer en pocos meses.
David Vetter era el tercer hijo de David Jr. y Carol Ann, un matrimonio del norte de Houston. Ellos habían tenido una hija saludable, Katherine, pero su segundo hijo nació con la inmunodeficiencia combinada grave y murió a los siete meses de vida. Cuando la pareja supo de su nuevo embarazo –el cual traería a David a este mundo–, se realizó las pruebas para determinar si nacería en las mismas condiciones. Lamentablemente, los estudios lo confirmaron. Los médicos, incapaces de dar una solución satisfactoria a la pareja, plantearon aislar al niño en una burbuja de plástico y mantenerlo lejos de cualquier agente infeccioso hasta que pudieran encontrar una cura. Esta medida –en principio, provisional– se convirtió en la vida de David Vetter. El niño creció sin poder recibir un beso de buenas noches de su madre, sin sentir el calor de un abrazo y sin poder sentir el rocío sobre su piel. Esta situación generó debates éticos en todos los campos, además de llevar a David a la fama y después al cine (imagen 1).

A diferencia de las películas, nuestra historia no tiene un final feliz. A los 12 años, el estado psicológico de David se comenzó a deteriorar de manera preocupante y los médicos decidieron intentar el último recurso, un trasplante de médula de su hermana Katherin. La operación pareció ser un éxito, pero unos meses después David Vetter presentó vómitos, fiebre, diarrea y sangrado intestinal. El diagnóstico fue linfoma de Burkitt, un tipo de leucemia producido por el virus de Epstein-Barr. ¿Cómo había contraído el virus? Los investigadores descubrieron que la médula que donó su hermana había sido la responsable de la infección. Y es que cerca del 90% de la población está infectada con el virus de Epstein-Barr, aunque nuestro sistema inmune lo mantiene a raya, pero en el cuerpo de David Vetter, sin sistema inmune que lo defienda, tuvo consecuencias devastadoras. David murió a las dos semanas, cerró los ojos unos minutos después de recibir el primer y único beso de su madre.
Una existencia sin contacto
La historia de David Vetter no es la única; miles de niños han tenido que pasar alguna parte de su vida aislados del mundo en una cámara. Su única esperanza era un trasplante de unas células que se encuentran en la médula y que son capaces de dar origen a los diferentes tipos de células de la sangre, como las del sistema inmune. Éstas son llamadas células hematopoyéticas progenitoras y gracias a los adelantos de la ciencia, la tasa de éxito para el trasplante es del 90%. Pero, como vimos en la historia de David Vetter, hay varios problemas que se deben superar, como: 1) conseguir un donador compatible, 2) valorar si hay infecciones y la gravedad de éstas, 3) realizar un diagnóstico temprano, y 4) realizar el tratamiento de preferencia antes de los cuatro meses de vida.
Uno de los problemas más recurrentes en cualquier trasplante de órganos es la compatibilidad del donador, ya que en muchos casos no se cuenta con un donador 100% compatible. Entonces se recurre a donadores haploidénticos, es decir, donadores que son compatibles con algunos de los genes que participan en el rechazo de órganos, pero no en todos [b]. El donador principal suele ser un hermano; en caso de que no lo haya, se recurre a un donador no relacionado pero compatible, donde se acostumbra utilizar células de cordón umbilical o de la sangre. Y cuando no hay ninguna de estas posibilidades, se puede optar por células de los padres, preferentemente de la madre.
El éxito del trasplante haploidéntico es limitado, principalmente por la disponibilidad de donadores compatibles y por la posibilidad del desarrollo de la enfermedad “injerto contra huésped”, una enfermedad en la que las células del sistema inmune del paciente reaccionan contra las células donadas. La enfermedad injerto contra huésped puede hacer pasar muy malos ratos si se desarrolla rápidamente o de manera crónica, después de 100 días del trasplante. La incidencia para la primera es de un 25% y para la segunda de un 16%. En la década de los 90, los trasplantes de donadores parcialmente compatibles se asociaron con tasas altas de mortalidad y morbilidad como resultado de la reacción injerto contra huésped.

El problema de los niños con inmunodeficiencia combinada grave se resume a que tienen células defectuosas del sistema inmune, las cuales hay que eliminar y sustituir por células sanas y compatibles, pero si en vez de sustituirlas pudiéramos repararlas, ¿acaso no solucionaría esto la búsqueda de donadores?
Aquí es donde entra la terapia génica. Podemos imaginar la terapia génica como un tratamiento para sanar las células enfermas al insertar genes sanos que cumplan las funciones de los genes defectuosos. El procedimiento consiste en lo siguiente: primero se extraen las células de la médula de los pacientes con inmunodeficiencia combinada grave; después, se llevan estas células al laboratorio donde se les introduce el gen con la secuencia correcta y, por último, se vuelven a introducir las células al paciente enfermo. Parece fácil, ¿verdad? El concepto es sencillo de entender, pero hay muchos problemas técnicos que los científicos tuvieron que superar.
Romper la burbuja
Uno de los problemas es que los genes que se deben introducir son moléculas muy pequeñas de unos 10 micrómetros de longitud por dos nanómetros de ancho. Para darnos una idea del minúsculo tamaño, si el gen tuviera el grosor de un cabello humano, nuestra mano tendría la altura del monte Everest. Entonces, ¿cómo podemos manipular algo tan pequeño como un gen para lograr introducirlo dentro de las células? Para esto los investigadores se valieron de los virus. Sí, esos patógenos que causan enfermedades como el temido ébola, el coronavirus o la influenza. Los virus son partículas capaces de transportar ADN e introducirlo a las células que infectan, por lo que podemos usarlos para llevar la carga que deseamos como los genes sanos que requieren las células de los pacientes con inmunodeficiencia combinada grave.
Antes de usar los virus en los tratamientos clínicos, los científicos eliminan su capacidad de reproducción y de causar enfermedades, así resultan inofensivos para el paciente. Después estos virus se cargan con los genes terapéuticos y se convierten en una excelente herramienta de entrega genética. En el laboratorio, los virus se introducen en las células enfermas del paciente y éstos se encargan de llevar la secuencia de ADN sano al núcleo e integrarla en el ADN de esas células. Las células sanas son transferidas nuevamente a los pacientes, donde proliferan y se diferencian para formar células del sistema inmune funcionales.
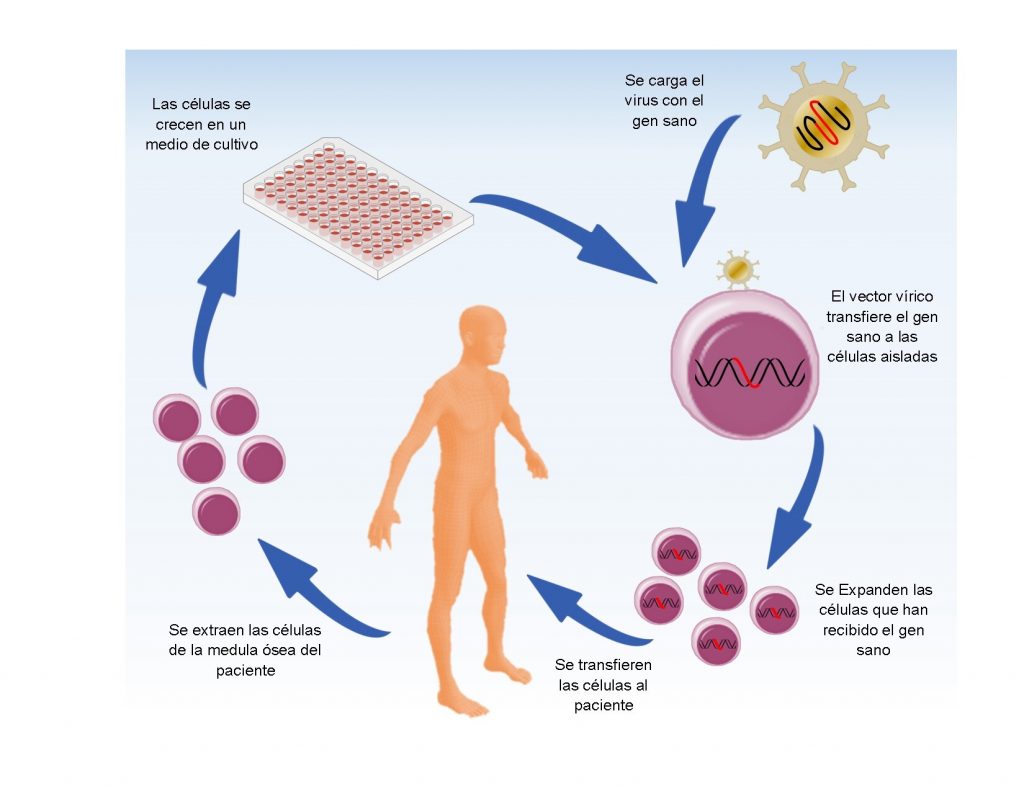
Los ensayos clínicos muestran que la terapia génica es igual o superior en efectividad que el trasplante haploidéntico en cuanto a restablecer el sistema inmunológico de los pacientes. Lo más importante es que no hay rechazo de tejido, ¡ya que son las mismas células del paciente! La larga carrera de buscar un donador compatible ha terminado.
La terapia génica ha tenido sus inconvenientes. Por ejemplo, en algunos pacientes se observó el desarrollo de leucemia linfoblástica aguda, un cáncer que afecta los glóbulos blancos de la sangre, esto debido a que el virus que se usó como vector para llevar el gen sano se insertó en un lugar inadecuado, cerca de un gen que favorece el desarrollo del cáncer –a este tipo de genes se les llama oncogenes–. A partir de esos inconvenientes se han desarrollado transportadores virales más seguros. En los pacientes tratados con estos nuevos transportadores virales no se ha observado el desarrollo de leucemias después de cuatro años del tratamiento.
Otro problema que se tuvo que sortear con la terapia génica fue que se conseguía únicamente la corrección de un tipo de células del sistema inmune, las células T. La cuestión era que el paciente ya tenía células defectuosas que ocupan los nichos para las células nuevas; como dicen: ¿cómo pondré más café en tu taza si ya está llena? Para favorecer el desarrollo de otros grupos de células los científicos eliminaron las células defectuosas del paciente por quimioterapia –un tratamiento conocido como mieloablación o acondicionamiento–. Esto permitió que las células corregidas por terapia génica pudieran sustituir a las células defectuosas.
La enfermedad del niño de la burbuja o inmunodeficiencia combinada grave fue sólo la primera condición en ser corregida con terapia génica. Actualmente, se están desarrollando terapias para más enfermedades y hay varios medicamentos basados en terapia génica viral disponibles en el mercado. Entre las enfermedades que los científicos están investigando está el síndrome de Wiskott-Aldrich, una enfermedad caracterizada por sangrado, infecciones, vasculitis o linfoma, y la enfermedad granulomatosa crónica XL, donde hay deficiencia de una proteína clave en la respuesta inmune, lo cual expone al paciente a infecciones e inflamación grave recurrente.
Una opción atractiva para aplicar en la terapia génica es la edición del genoma usando las tijeras moleculares (CRISPR/Cas9), una tecnología que permite a los científicos insertar secuencias de ADN en regiones específicas, y que le valió el Premio Nobel de química en 2020 a sus descubridoras Jennifer Doudna y Emmanuelle Charpentier. Esta técnica usa proteínas procedentes de bacterias para dirigir la inserción de ADN en regiones específicas, lo que permitiría colocar las secuencias de ADN en las regiones adecuadas. La aplicación de esta técnica al tratamiento de la inmunodeficiencia combinada grave está todavía en fase de experimentación. Hablaremos más a profundidad sobre las tijeras moleculares en un próximo artículo. Por lo tanto, todavía queda un camino largo por recorrer para brindar a la sociedad terapias eficaces y seguras, pero los avances en la ciencia son innegables y si David Vetter hubiera contado con los tratamientos actuales, su vida hubiera sido totalmente diferente.
Referencias
Fischer, A. & Hacein-Bey-Abina, S. (2020) Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med, 217 (2): e20190607. doi: https://doi.org/10.1084/jem.20190607
Shirley, J. L., de Jong, Y. P., Terhorst, C., Herzog, R. W. (2020). Immune Responses to Viral Gene Therapy Vectors. Molecular therapy: the journal of the American Society of Gene Therapy, 28(3), 709–722. https://doi.org/10.1016/j.ymthe.2020.01.001
Para saber más
[a] La inmunodeficiencia combinada grave se debe a mutaciones en el ADN, es decir, variaciones en la secuencia de moléculas que llevan la información para sintetizar las proteínas de nuestro cuerpo. Estas mutaciones se han identificado en 17 genes que participan en la síntesis de proteínas del sistema inmunitario y que son cruciales para el desarrollo de linfocitos T, linfocitos B y linfocitos Natural Killer, lo que lleva a un sistema inmune defectuoso y a la muerte del paciente en pocos meses a causa de infecciones.
[b] Las células del sistema inmune no sólo reconocen patógenos, si no también pueden reconocer células de un trasplante por la expresión en su superficie de proteínas llamadas antígenos leucocitarios humanos (HLA), de las cuales existen muchos tipos diferentes y las personas difieren en las moléculas de HLA que expresan. Por lo que al buscar un donador para un trasplante sus moléculas del HLA deben ser lo más similares posibles. Cuando hay similitud parcial en las moléculas de HLA entre el donante y el receptor el trasplante se llama haploidéntico.




Vórtice, enero-mayo 2021 es una publicación trimestral digital editada por la Universidad Autónoma del Estado de Morelos (UAEM), a través de la Dirección de Publicaciones y Divulgación, Edificio 59 (Facultad de Artes), Campus Norte. Av. Universidad 1001, Col. Chamilpa, CP 62209, Cuernavaca, Morelos, México. Teléfono +52 777 329 7000, ext. 3815. Correo: revistavortice@uaem.mx. Editora responsable: Jade Gutiérrez Hardt. Reserva de Derechos al Uso Exclusivo No. 04-2014-070112203700-203, ISSN 2395-8871, ambos otorgados por el Instituto Nacional del Derecho de Autor.
Responsable de la última actualización de este número: Roberto Abad, Av. Universidad 1001, Col. Chamilpa, CP 62209.
Vórtice está incluida en el Índice de Revistas Mexicanas de Divulgación Científica y Tecnológica del Consejo Nacional de Ciencia y Tecnología (Conacyt). Publica artículos de divulgación relacionados con las ciencias y las humanidades, y textos breves que transmitan el gusto por el conocimiento científico. El contenido de los artículos es responsabilidad de cada autor. Esta revista proporciona acceso abierto inmediato a su contenido, con base en el principio de ofrecer al público un acceso libre a las investigaciones para contribuir a un mayor intercambio global de conocimientos. Se distribuye bajo una licencia Creative Commons Reconocimiento-NoComercial 4.0 Internacional License.